Waar je in Nederland als app-ontwikkelaar te maken hebt met de Inspectie voor de Gezondheidszorg, worden medische apps in Amerika door de FDA gecontroleerd. Deze Food and Drug Administration is een agentschap van de federale overheid dat zich bezighoudt met de kwaliteit en veiligheid van onder andere voedsel, cosmetica, medicijnen en medische producten. In 2013 bracht de FDA een lang-verwachte “vrijblijvende en verklarende” richtlijn uit voor medische app-ontwikkelaars. Hoe staat het daar inmiddels mee?
De FDA houdt zich al jaren bezig met het reguleren van software van medische hulpmiddelen – ook op mobiele platformen. In 1989 kwam de FDA voor het eerst naar buiten met een standpunt over hoe ze van plan waren te bepalen of een product gebaseerd op een computer of op software een hulpmiddel is – en of de FDA zo’n hulpmiddel in dat geval moet reguleren. Na 1989 groeide het gebruik van computer- en software producten exponentieel; de producten werden steeds complexer en er kwamen er steeds meer medische hulpmiddelen, waaronder software, bij. Lange tijd hanteerde de FDA dezelfde wet- en regelgeving voor mobiele medische apps die ook voor bestaande medische hulpmiddelen gold.
Met exponentieel toenemende technologie voldoet de wet uit 1989 niet langer
Maar door de explosieve groei van medische apps, die lang niet allemaal strikt als medisch hulpmiddel gebruikt worden, en integratie van mobiele netwerken en diensten in bestaande medische technologie verandert het Amerikaanse zorglandschap in rap tempo. Pas in 2011 brengt de FDA een eerste klad-versie van richtlijnen (guidance) voor medische apps uit. De definitieve versie (final guidance) volgt pas twee jaar later, in september 2013. In de tussentijd volgen er hoorzittingen en besprekingen in het Congres over de rol die de overheid zou moeten spelen bij de opkomende markt van medische software en apps als medische hulpmiddelen. Met het uitblijven van een officieel standpunt van de FDA rondom medische software opereren Amerikaanse mHealth ondernemers zodoende lange tijd in een grijs gebied.

Met AliveCor wordt een smartphone een hartritme-monitor: FDA approved
Waarom heeft het ruim 2 jaar gekost om met een definitieve richtlijn te komen? Erik Vollebregt, advocaat en partner bij Axon en gespecialiseerd in wetgeving rond medische hulpmiddelen en technologie, verklaart: "Amerika kent een sterke lobby van bedrijven die van mening zijn dat de FDA niets over dit onderwerp te zeggen moet hebben. Die lobby is een mengeling van
"Software-ontwikkelaars willen graag buiten dat lange en kostbare traject blijven"
gevestigde grote spelers - die meer middelen hebben om te lobbyen - en kleine ondernemers, die het argument delen dat niet alle medische apps een hoog risico vormen. Nadat de eerste draft guidance werd gepubliceerd is er veel commentaar geleverd vanuit de industrie op de FDA. Het toelatingsproces voor een FDA clearance is een gecompliceerd traject en duurt lang, dus software-ontwikkelaars willen graag buiten dat traject vallen."
Vollebregt: "Ook in Europa gold in 2007 de vraag of stand-alone software (zoals apps) onder de definitie van medische hulpmiddelen valt, en was die definitie niet volledig eenduidig. De materiële scope van medische apps - wat de wetgever beschouwt als medisch hulpmiddel - komt redelijk overeen tussen de VS en Europa. Maar in tegenstelling tot Europa - waar een soortgelijke discussie wordt gevoerd over wetgeving rondom software als medisch hulpmiddel - is de discussie in Amerika heel politiek geworden."
Dinosaurus
Dat debat over de regulerende rol van de FDA kent voor- en tegenstanders. Voorstanders geven aan dat de FDA niet stil heeft gezeten in de afgelopen jaren: meer dan 100 medische apps zijn gereguleerd en goedgekeurd door het agentschap, hoewel een officieel standpunt over regulerende rol van de FDA nog geformuleerd moest worden. Maar de tegenstanders, specifiek vanuit de mobile health achterban, zijn er van overtuigd dat te veel controle vanuit de FDA, de verwarrende regels en bureaucratie een rem zetten op innovatie. Waarom zou een dinosaurus als de FDA zich bezig houden met de nieuwste mHealth technologieën, is het sentiment bij health tech ondernemers en lobbyisten.
Waarom zou een dinosaurus als de FDA zich buigen over de nieuwste mHealth technologieën?
Er ligt inmiddels een final guidance - een richtlijn waar ondernemers, consumenten en de overheid zich op kunnen baseren bij gebruik van medische apps. De definitie van een app als medisch hulpmiddel die hierin wordt gebruikt is: een app die bedoeld is voor het diagnosticeren van een ziekte of andere conditie, of het genezen, verzachten, behandelen of voorkomen van een ziekte, or is intended to affect the structure or any function of the body of man.

Die richtlijn maakt voor software-ontwikkelaars en ondernemers duidelijk wat de FDA beschouwt als ‘mobiele medische apps’, en waar het agentschap het zwaartepunt legt wat controle betreft. Want de mobiele medische apps die de FDA reguleert hebben als kenmerk dat ze gebruikt worden in combinatie met een reeds bestaand en gecontroleerd medisch hulpmiddel, of dat ze het mobiele platform (de tablet of smartphone) tot
'Mobiele medische apps' ondersteunen traditionele medische hulpmiddelen, of werken stand-alone
een medisch hulpmiddel maken. Daarmee kan gedacht worden aan een app die een traditioneel medische hulpmiddel ondersteunt (een iPad app die een dokter helpt om een geïmplanteerde pacemaker te beheren) en apps die stand-alone (zelfstandig) werken, bijvoorbeeld een app voor radiologen om medische beelden te bekijken op een tablet, waar ook algoritmes in verwerkt zitten om potentieel schadelijk weefsel te herkennen.
Kortom, de FDA richt zich daarmee op een klein gedeelte van het grote aanbod van medische apps, specifiek op hoge risico-klassen, en richt zich op functionaliteit in plaats van platform. Met die op risico-gebaseerde aanpak hoopt de overheidsorganisatie patiënten te kunnen beschermen voor risicovolle applicaties, en tegelijkertijd ruimte te kunnen bieden voor innovatie en nieuwe mHealth producten.
Glucose-meter of slaap-apneu
Het agentschap neemt een wat passievere houding aan wat betreft apps die een laag risico vormen. De FDA schrijft dat mobiele apps die patiënten gebruiken bij het managen van hun ziekte, uitleg geven over ziektes en behandelingen, of bijvoorbeeld toegang geven tot elektronische patientendossiers geven wel in de categorie ‘medisch’ kunnen vallen, maar een laag risico vormen voor patiënten en daarom niet top priority zijn. Maar de FDA behoudt zich door de zogenaamde enforcement discretion, opgenomen in de richtlijn, ook het recht om zaken die niet expliciet genoemd worden later te kunnen handhaven, als een slag om de arm in een markt die volop in ontwikkeling is.
Inmiddels heeft het agentschap al zo’n 100 medische apps beoordeeld
Medische apps die door de FDA worden gekeurd, zullen aan dezelfde standaarden en eisen moeten voldoen als alle andere medische hulpmiddelen. Inmiddels heeft het agentschap al zo’n 100 medische apps beoordeeld, van externe ECG-apparaten tot glucosemeters en smartphone echo’s. Sommige van die gereguleerde apps zijn op consumenten gericht, zoals een glucose testsysteem voor thuis, of een slaap--apneu monitor. Maar de FDA controleert ook medische hulpmiddelen voor zorgprofessionals.
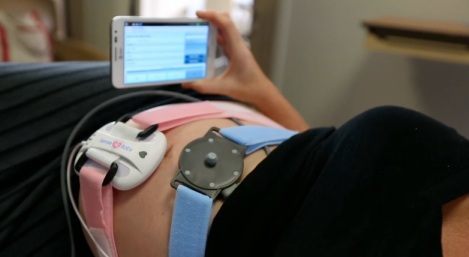
Sense4Baby biedt thuismonitoring met draagbare apparatuur en een tablet
Het bedrijf Telenatal bracht een Amerikaanse innovatie op de Nederlandse markt en was nauw betrokken bij het reguleringproces van de FDA. Met Sense4Baby, bedoeld zwangere vrouwen die medisch risico lopen met behulp van een tablet en draagbaar CTG-apparaat thuis te monitoren, kreeg ondernemer Chris Kerkhof te maken met Amerikaanse wet- en regelgeving. "We zijn ruim een jaar bezig geweest met FDA approval voor de technologie en software, en vier maanden met Europese CE-markering." Het veelgehoorde argument dat FDA goedkeuring een gecompliceerd en lang traject is, lijkt daarmee te worden bevestigd. Kerkhof: "Het meesturen van klinische resultaten van het CE-certificeringsproces bespoedigde de FDA procedure."
"Het FDA-traject duurde een jaar, de CE-markering was in 4 maanden rond"
Kerkhof herkent de juridische cultuur in de VS. "Nadat Sense4Baby ruim drie jaar geleden werd geïntroduceerd in de VS, bleek er nog geen toepassing voor de praktijk te zijn. Nederland kent een lange traditie van thuismonitoring, dus daarom werd de innovatie hier geïntroduceerd. Met een cultuur van procederen zijn artsen bang dat er iets met de patiënt gebeurt wanneer er buiten de kliniek of het ziekenhuis met nieuwe technologie of software gewerkt wordt, ook al zijn Amerikaanse artsen enthousiast." De FDA approval en CE-markering geven in ieder geval de mogelijkheid om de mHealth-innovatie aan beide kanten van de oceaan op de markt te brengen.
Waarschuwing
Hoewel de definitieve richtlijn nog volop werd bediscussieerd in het Congres, zit de FDA niet bepaald stil. In mei 2013 onderzocht het agentschap een app die niet goedgekeurd was, maar wel als medisch hulpmiddel beschouwd werd. Het ging om een applicatie genaamd uCheck, die urine teststrips met behulp van een smartphone controleert. Met de ingebouwde camera van een iPhone checkt de app de concentraties van bepaalde stoffen in de teststrip, die kunnen duiden op urineweginfecties. De brief van de FDA aan Biosense Technologies, de makers van uCheck, is een waarschuwing voor andere ontwikkelaars. Maar over het algemeen is de overheidsinstantie in 2013 voorzichtig geweest met het controleren en waarschuwen van software-ontwikkelaars en ondernemers.
Balancerend tussen de belangen van ondernemers en de veiligheid van consumenten heeft de FDA geen gemakkelijke positie. Bovendien maakt de sterke lobby en politieke discussie het niet gemakkelijk voor de overheidsinstantie om wetgeving te vormen rond het thema mHealth. Maar de final guidance biedt een - lang verwachte - richtlijn die meer duidelijkheid geeft over software als medisch hulpmiddel. Hoewel de richtlijn geen wet of wetgevend document is, biedt deze wel meer duiding en zekerheid aan ontwikkelaars die lange tijd in relatieve onzekerheid werkten.





Wat is de final guidance dan precies?